The FDA Q-Submission Process: A Medtech founder’s guide to the Pre-Sub


TL;DR
A Q-Submission (most often a Pre-Sub) is a free, formal way to get FDA’s written feedback on your device and your submission plan before you file. You send a short package with specific questions, FDA responds in writing within about 75 days, and they usually offer a meeting to talk it through. Done well, it can save you 6 to 18 months of building the wrong submission. Here’s what it is, when to file, what to ask, the four mistakes I see founders make most, and what goes in the package.
The FDA’s open invitation founders hesitate to take
Ever heard of a Q-Submission? More founders these days know this little feedback mechanism exists, but most don’t know how to use it properly.
Here’s what it does. It’s how the FDA tells you what they think of your plan before you spend nine months building the wrong submission package and sending it in. Most teams only find out their plan was off after they file, when the deficiency letter lands. A Q-Sub moves that conversation to the front, where it’s cheap to act on.
It’s also free. You can ask the FDA anything reasonable about your device, your testing, your strategy, or your pathway, and they’ll answer in writing within 75 days. No fee. Technically the application form is no longer than a few pages. The only thing it costs you is the time to put the questions together properly and the attachments you submit along with the form.
Most teams we work with have never done one. By the time they reach us, half are assuming they don’t need it because their pathway “is obvious.” The other half assume the feedback won’t be worth the effort and time to wait. Both groups are usually wrong.
I think the hesitation comes from how founders picture the FDA. They imagine a black box, or worse, an adversary waiting to say no. The reality is closer to the opposite. FDA built a front door, labelled it, and told everyone it’s free to walk through. Most founders just walk past it on the way to spending a year and a small fortune building a submission nobody at the agency has ever seen.
Let me walk through what a Q-Submission actually is, when to do one, what to ask the FDA, and the four mistakes I see founders make most often.
What a Q-Submission actually is
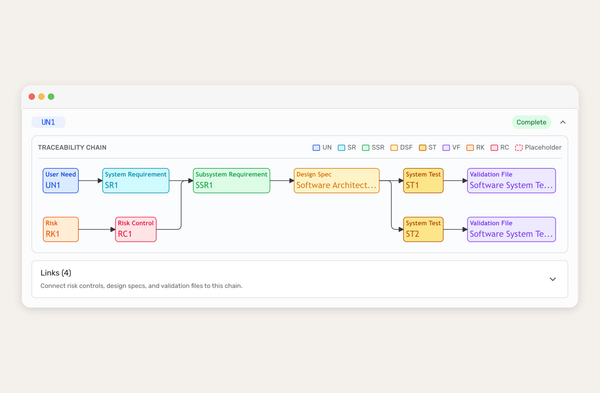
A Q-Submission is a request for written FDA feedback. A pre-submission is a request for feedback related to a future submission. It’s the formal mechanism FDA created to let device makers de-risk a submission before they file it.
FDA’s guidance document on the Q-Sub program has the full mechanics: Requests for Feedback and Meetings for Medical Device Submissions (Q-Submission Program).
In practicality, you submit a short package describing your device, the submission you’re planning (510(k), De Novo, PMA, IDE), and a list of specific questions you want the FDA to answer. The FDA reviews it, sends back written feedback, and usually offers a meeting to discuss that feedback live.
The output is FDA’s actual position on your specific device. Not a guess or Reddit post, but their position in writing, that you (and they) can reference in your eventual submission.
That last part matters more than it sounds. Review teams rotate, memories fade, and a friendly phone call evaporates the moment a submission gets reassigned. A written Pre-Sub response is something that FDA will have access to a year later and say: here is what FDA told us, and here is how we built to it. That changes the tone of a review.
Side note: Pre-submissions are subject to FOIA requests, so don’t put any trade secrets in there.
The four most common Q-Sub types you should know
There’s more than one kind of Q-Submission. Most founders only know about the Pre-Sub.
- Pre-Submission (Pre-Sub). The most common one. You’re planning a future submission and you want FDA’s feedback on your strategy, testing plan, predicate, or specific technical questions. Most founders should start here.
- Submission Issue Request (SIR). You already have a submission with FDA, FDA has flagged an issue, and you want to discuss it before you formally respond. Less common at the founder stage.
- Study Risk Determination (SRD). You’re running a clinical study and you need FDA’s view on whether it qualifies as a Significant Risk or Non-Significant Risk study. Specific to clinical-data work.
- Informational Meeting. Pure information sharing, no specific submission planned. Useful for novel technology where you want to start a relationship with FDA before you’re ready to formally engage.
There are actually a few more, like some tied to the PMA process, but these are the most common. If you’re a founder reading this and you’re not sure which one applies, it’s almost always the Pre-Sub. There are a few additional types, like Accessory Classifications and PMA 100 Day Meetings, mostly for unique requests.
When to file a Pre-Sub
The biggest mistake on timing isn’t filing too late. It’s filing too early.
If you file a Pre-Sub before you have a clear device design, a defined intended use, and at least a preliminary testing plan, the FDA will tell you they need more information to give you useful feedback. Just because a Pre-Sub is free, doesn’t mean you shouldn’t spend meaningful amount of time preparing it.
You’ll wait 75 days to hear “we need more information.” That isn’t FDA being unhelpful. It's the FDA telling you, accurately, that you don’t have enough specificity for them to engage meaningfully.
The sweet spot for a Pre-Sub is when you have:
- A clear intended use statement
- A target FDA product code (or a reasonable hypothesis if you’re in De Novo territory)
- A high level strategy for your testing plan (including numbers and locations if possible)
- A predicate device candidate (for the 510(k) pathway) or a clear classification rationale
- Specific questions where FDA’s feedback would change something concrete in your plan
That sweet spot usually lands somewhere between your design lock and your verification testing. For most teams that’s six to twelve months before you’d otherwise file the formal submission. Earlier than that and you don’t have the specificity. Later than that and you’ve already invested in testing, the FDA might want you to redesign.
Here’s what too early looks like in practice. A founder has a clever idea and a rough prototype, has multiple intended uses they could pursue with no fixed materials or design and they want FDA to bless the concept. FDA can’t. There’s nothing concrete to react to. Wait until your design is solidified enough that FDA’s answer would actually change what you do next. FDA aren’t there to workshop with you, they’re there to say yes, no, and why.
The four mistakes I see most often
The questions are where most Pre-Subs succeed or fail. Ask the right ones and you’ll come away with FDA-aligned guidance you can build the rest of your submission around. Ask the wrong ones and you’ll get back a polite document that didn’t move you forward.
After running Pre-Subs across our client work, the same four founder mistakes show up over and over.
1. Asking questions the FDA won’t answer in a Pre-Sub.
The FDA explicitly lists what it will and won’t address in a Pre-Sub. They will not tell you what your device classification is in a Pre-Sub. That’s what a 513(g) is for. They won’t tell you whether your device is regulated at all. That’s what a Request for Designation is for. Asking FDA in a Pre-Sub for things they’ve publicly said they won’t answer is a wasted question. They’ll point you to the right pathway, and your real questions get fewer pages of attention.
2. Not giving the FDA enough information to answer the question.
If you’re asking about a clinical trial, send the protocol. If you’re asking about biocompatibility testing, send the device materials breakdown. There is nothing worse than waiting 75 days only to find out the FDA couldn’t answer half your questions because they didn’t have enough to go on. This one is almost always self-inflicted. The founder didn’t want to write a long package, the FDA didn’t have what it needed, and the whole thing goes nowhere. Provide an attachment for most of your questions, it's worth it.
3. Asking the FDA open-ended strategy questions.
“Should we run a clinical trial?” is the founder version of asking a child if they want candy. Of course the answer is yes. Asking FDA open-ended “what would you recommend” questions invites them to recommend the most conservative possible approach. That isn’t the FDA being difficult. It's the FDA doing its job.
The fix: have a plan and have a rationale. I always say, don’t ask the FDA to write your strategy, ask them whether they agree with the one you’ve already written. If they don’t, they’ll tell you why. For example, if you think biocompatibility doesn’t apply to your materials, don’t just say it's “because our materials are proven to be safe”. Say “because x, y, and z are listed on Attachment G of the FDA guidance on biocompatibility which identifies as materials excluded from ISO 10993-1 testing”. Shows you’ve done your homework and is actionable for FDA.
4. Raising integrity questions in the meeting, or ignoring the FDA’s advice in later submissions.
A Pre-Sub is your first formal interaction with the FDA on this device. It’s also the FDA’s first formal look at you. You don’t want to give them a reason to question your integrity. That doesn’t mean it's submissive. It means be clear-eyed and serious. If the FDA gives you advice in a Pre-Sub and you don’t take it in the actual submission, expect them to ask why, and expect a tougher review. Don't argue with them to argue and don't give them reasons to be suspicious about what you're doing. FDA are particularly to you marketing your "FDA cleared" product before even submitting anything to them. Don't do that. They check your website.
5. Including information not relevant to your pre-submission
The opposite of point 2 above is true. If you are not asking any questions on usability testing, you don't need to spend a ton of time working out your usability evaluation protocol for them. Seems obvious but you can run multiple pre-submission sequentially so scope the info you submit to your specific questions so FDA can answer them.
This only becomes difficult because it forces you to solidify your strategy and your product scope, which isn't always easy in early days. While it may take some time, we found with our clients it's absolutely worth spending time on your Pre-Sub package at this stage.
What goes in a Pre-Sub package
The package is shorter than most founders expect. The standard package includes:
- preSTAR PDF. This is FDA's preferred format for submitting these days. It is a structured PDF walking you through the details to include. Not everything is required for submission so scope the info towards your question.
- Cover letter. One page that identifies the submission, the device, the planned future submission type, and the questions you plan to ask.
- Comprehensive device description. Five to fifteen pages on what the device is, what it does, materials, technology, indications for use, intended use, target user. Focused on the parts FDA needs to answer your questions.
- Specific questions. Each question listed out along with the supporting context for why you are asking it. Usually a few sentences long, your questions should cover maximum of 4 categories and I recommend no more than 8 questions maximum. Asking too many unrelated questions can make FDAs review difficult and they may ask you to submit another pre-submission.
- Supporting attachments. The preSTAR allows for attachments Whatever the FDA needs to answer the questions. Testing protocols, draft labeling, predicate comparison tables, software documentation level rationale, biocompatibility planning, whatever applies.
The whole package can take anywhere from weeks to months to put together depending on how industrious your team is. Always ask for a meeting and not just written feedback. The meeting is an opportunity to discuss anything from their written responses you want feedback on.
Cost and timeline
The Pre-Sub itself is free. There is no FDA user fee for a Q-Submission of any type. The cost is your time, any consulting help you bring in, and the 60 to 75 days FDA takes to review and respond.
The 75-day clock starts the day the FDA receives your package. They’ll send a feedback letter with their official responses to your questions. The meeting is a 60-minute call. Bring your team, walk them through the questions, listen to their responses carefully, take notes, and don't be afraid to ask for clarification. The verbal feedback in the meeting often goes deeper than the written letter. That’s where you learn what the FDA is actually thinking.
How Formly accelerates this
This is where I’ll be direct about why we exist. The Pre-Sub process is one of the highest-leverage moments in the entire FDA pathway. Done right, a Pre-Sub saves you 6 to 18 months of submission risk. Done wrong, it wastes 75 days and gives you a feedback letter that changes nothing.
Most of our engagements start with a Pre-Sub. We frame the scope, draft the questions, build the package with attachments, attend the meeting, and translate the FDA’s feedback into a concrete adjustment to your strategy. The model is expert-led and AI-enabled. Senior regulatory people own the judgment calls, and our systems make the documentation reliably fast. The output is FDA-aligned guidance you can take into your submission with confidence. We consistently turn pre-submissions around in under a few weeks.
The translation step is the part founders underrate. The FDA’s feedback is rarely a clean yes or no. It’s a set of signals about what they’re worried about, and the work is turning those signals into specific changes to your test plan, your predicate choice, or your intended use, before they harden into a deficiency letter on the real submission. If they "highly recommend" you do something, you better do it.
If you’re a founder who has never written a Pre-Sub, I’d strongly recommend not making your first one your only one. The learning curve is steep, and the cost of getting it wrong is real.
A few things to keep in mind
- File at the right moment. After design lock, before you’ve sunk cost into verification testing.
- Ask bounded questions the FDA can actually answer. Confirm or correct, don’t attempt to outsource your strategy to them.
- Send enough information. If you reference a protocol or a materials breakdown, include it.
- Treat the meeting as the real prize. The verbal feedback usually goes deeper than the letter.
- Take the FDA’s advice. If you decide not to, be ready to explain why at submission.
Prepping a Pre-Sub? We built a program for exactly this
Everything above is doable on your own. It's also easy to get wrong the first time, and the first time is an expensive place to learn. So we built the FDA Pathway & Pre-Submission Meeting Program to get founders from wherever they're standing to a Q-Sub meeting they can actually use. There's no fixed sequence, because founders don't show up at the same place.
Some have already filed and need a strategy for the FDA's questions. Some have a package drafted and want it stress-tested before it goes in. Some are starting from scratch and don't yet know what to ask. We meet you where you are and help do the thinking that turns your position into a productive FDA conversation. You walk in knowing exactly what you're asking and why, and you walk out with the FDA's position translated into concrete next steps, not a feedback letter you're left to decode alone.
We open three spots a month, at no cost. The regulatory system isn't built to be kind to early founders, and running the program for free is one small way we try to even that out.
Apply for the FDA Pathway & Pre-Submission Meeting Program.
Entering the EU? We've got you covered there too
Heading to the EU as well, or instead? The system works nothing like the FDA. There's no single agency and no Pre-Sub. Under the MDR, your gatekeeper is a Notified Body. A private, accredited organisation that reviews your technical file, audits your quality system, and issues your CE certificate.
The FDA is one front door. The EU is a corridor of private ones, and the part most founders miss is that which Notified Body you pick materially changes your timeline and your outcome. That's why we built the EU counterpart, the MDR Pathway & Notified Body Match program. We take three founders a month, through three crucial steps: a Pathway Assessment that ends with a one-page route-to-market summary, a Notified Body Match (the step founders underrate), and a Notified Body Intro Meeting we arrange and sit in on with you. All at no cost.
Apply for the MDR pathway & Notified Body match program.
Spencer Todd is the CEO and co-founder of Formly, a regulatory partner for medical device companies. Before founding Formly, he spent years at the FDA and as a medical device consultant.